|
Introduction The chemistry of 1,3,4–oxadiazoles has attracted a good number of experimental and theoretical studies in recent days [1-4]. These molecules show antimicrobial, anti-inflammatory and insecticidal properties [5-8]. There are extensive recent research reports highlighting the potential of the transition metal complexes of 1,3,4–oxadiazole dyes, photosensitive materials, liquid crystals, and inorganic light-emitting diodes [9-15]. These complexes were reported to have high electron affinities and found to have applications in OLED. Cobalt- and nickel-based transition metal complexes were explored as artificial photosynthetic systems for splitting water [16]. In natural photosynthetic systems, short-strong hydrogen bonds network with water to function as a proton exit channel [17]. These reports revealed that the Ni(II) and Co(II) derivatives of 1,3,4–oxadiazole with short strong hydrogen bonds will be effective for the development of next generation noble metal free artificial photosynthetic systems capable of splitting water. Considering the biological applications, properties suitable for developing new materials with special characteristics of 1,3,4-oxadizoles, the present work explores the electronic, spectral and orbital characteristics of Co(II) and Ni(II) complexes of 5-(pyridin-4-yl)–3H-1,3,4–oxadiazole-2–thione in addition to the presence of short-strong hydrogen bonds. Experimental Preparation method for the 5-(pyridin-4-yl)–3H-1,3,4–oxadiazole-2–thione ligand 5-(Pyridin-4-yl)-3H-1,3,4-oxadiazol-2-thione was prepared by cyclization of isonicotinic acid hydrazide using carbon disulphide in presence of KOH [18]. Yield: 70%, M.P: 2480C, MS: m/z = 179(M+), m/z = 180(MH+). All chemicals used were of Analar grade. Synthesis of the Ni(II) and Co(II) complexes of 5-(pyridin-4-yl)–3H-1,3,4–oxadiazole-2–thione analar grade chemicals were used for the synthesis of all the complexes. The Co(II) and Ni(II) complexes were prepared by adding a hot ethanolic solution of respective metal chlorides to a refluxing solution of the ligand in ethanol, maintaining the stoichiometric metal ligand ratio. The solid complexes separated out and were filtered off, washed with hot ethanol and dried (Yield 65-75%). The complexes are insoluble in all common organic solvents except in hot DMF. The melting point of the complexes were very high (>3000C). The melting point of the ligand is 2480C. The Co(II) complex turned a brown color, while the Ni(II) complex became pale green. Characterization Techniques The C, H, N contents were determined micro analytically. IR spectra were recorded on a Perkin-Elmer 1600 FTIR spectrophotometer. Molar conductance of the complexes in 1x10-3 mol dm-3 in DMF solution were measured using Systronics microprocessor based conductivity meter at room temperature. Magnetic susceptibilities were determined with Gouy assembly at room temperature, using Hg[Co(NCS)4] as the callibrant. 1HNMR (DMSO-d6) on Perkin-Elmer R-32 spectrometer at 90 MHz. Computational Methods The ground state geometries of the ligand and complexes were fully optimized at DFT level using Becke's 3 parameter density functional in combination with the non-local correlation functional of Lee, Yang, and Parr (B3LYP). 6-31+G basis sets were employed for the ligand and a relativistic effective core potential of Los Alamos and Double-ζ basis sets were employed for the metals (LANL2DZ). Vibration frequency calculations were performed on the optimized geometry to verify that these molecules are minima on the potential energy surfaces. Time dependent (TDDFT) calculations were performed to obtain the vertical singlet and triplet excitation energies. Natural population analysis (NPA) and natural bond orbital (NBO) analysis [19] were performed to obtain the nature of metal-ligand bond and cation-anion interactions. All the calculations were performed using the Gaussian-09 suite of program [20].Results & Discussion Geometry Analysis of Co(II) / Ni(II) Complexes The stoichiometries of the Co(II)/Ni(II) complexes of 5-(pyridine-4-yl)-3H-1,3,4-oxadiazole-2-thione were predicted by elemental analysis and molar conductance data included in Table 1. Accordingly the stoichimetries of the Co and Ni were predicted as [CoL2(H2O)2].4H2O and [NiL2(H2O)2].6H2O. The insolubility of the complexes and their low molar conductivities in DMF indicate that these complexes are non-electrolytic and polymeric in nature [21]. Therefore, a ligand may form a complex with a metal ion with S and another with N, which indicates that there will be four ligands around Ni and Co. The observed magnetic moments of cobalt and nickel complexes are 4.87 and 3.12 BM, respectively. These results evidenced the octahedral geometry of the complexes [22-23]. The electronic spectral bands observed for the Co(II) complex at 8860 cm-1, 17750 cm-1 and 19720 cm-1 are assigned to ν1[4T1g(F) → 4T2g(F)], ν2[4T1g(F)→4A2g(F)] and ν3[4T1g(F)→4T1g(P) transitions, respectively. The ligand field parameters calculated to be 838cm-1 (B), 942cm-1(Dq), 0.742(β) and 2.002(v2/v1). The Ni(II) complex exhibits three absorption bands at 9230cm-1,15050cm-1 and 25260cm-1. These lines are assigned to ν1[3A2g→3T2g], ν2[3A2g→3T1g(F)] and ν3[3A2g→ 3T1g(P)] transitions, respectively in the octahedral geometry [24]. The ligand field parameters of B=839 cm-1, Dq= 923cm-1, β = 0.685 and v2/v1 value of 1.63. The results of electronic spectral analysis also gave further evidence for octahedral geometry. Considering the results of magnetic moments analysis and electronic spectral analysis of the complexes, it is confirmed that the Co(II) and Ni(II) complexes possess octahedral geometry. The 1HNMR spectrum of the ligand HL recorded in DMSO-d6 showed two doublets centered at δ 8.79 (HC-N-CH) and δ 7.77 (HC-C-CH), indicating the involvement of oxadiazole and aromatic ring in complexation.
Evidence for Presence of Lattice Water
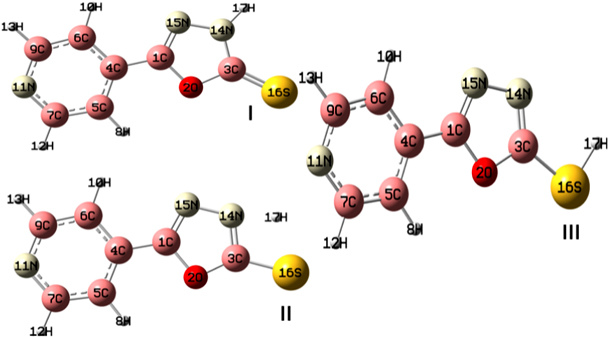
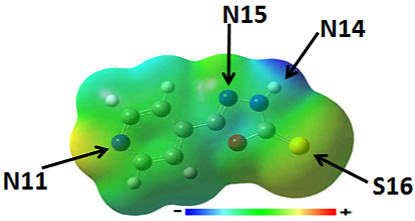
Optimization of Stable Geometries of Co(II) / Ni(II) Complexes Literature reports revealed that the 5-substituted-1,3,5-oxadiazole-2-thione may exist in two tautomeric forms [24]; the shifting of C=S stretching bands to 1200 cm-1 in our experimental IR spectrum also supports this observation. DFT calculations were performed with B3LYP/aug-cc-PVDZ level of theory in order to select the stable tautomer of the 5-substituted-1,3,5-oxadiazole-2-thione. The optimized structures of the two tautomeric forms along with the isolated transition states were included in Figure 1. The results of the DFT calculations revealed that the thione form shown in Figure 1(I) is more stable 10.3kcal/mol compared to the thiol form shown in Figure 1(II).The transition state isolated using QST2 possess an energy barrier of 34.6kcal/mol. Based on these results, the thione form of 5-substituted-1,3,5-oxadaiazole-2-thione is selected as the ligand for further study. The 5-substituted-1,3,5-oxadiazole-2-thione contains many potential coordination sites. The MESP is mapped on total electron density of the ligand by using ESP computations in order to optimize the active site of coordination on 5-substituted-1,3,5-oxadiazole-2-thione. When MESP is mapped on total electron density (Figure 2) of the ligand, the nitrogen of the six membered ring (indicated as N11 in Figure 2) and sulphur (indicated as S16 in Figure 3) are more active compared to nitrogen atoms of the oxadiazole ring. The absence of N-H and S-H vibration bands near 3500 cm-1 to 2500cm-1 in the experimental IR spectral pattern confirmed this observation. Accordingly, the nitrogen (N11) and sulphur (S16) of the six membered ring are selected as the active sites for coordination. The stoichiometric characteristics, magnetic moments evaluation and electronic spectral characteristics revealed that the Co(II) and Ni(II) complexes possess octahedral geometry. Different possible geometries for the metal complexes are optimized for all the geometries at B3LYP level.
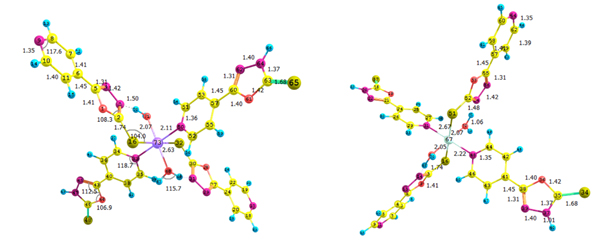
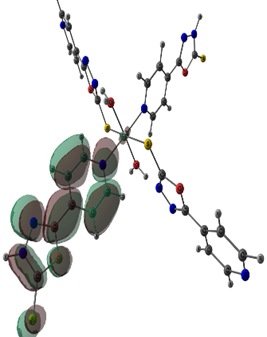
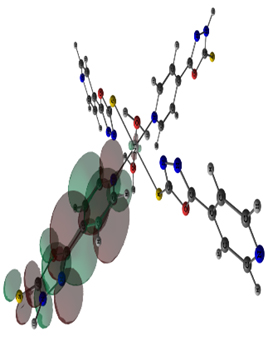
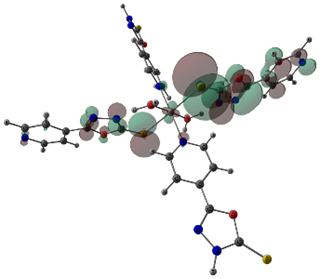
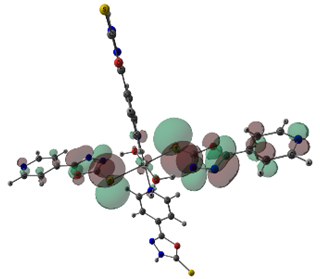
The stable geometries with no imaginary frequencies are given in Figure 3. The important optimized geometry parameters of the complexes are shown in Figure 3. The distances between the central cation and the nearest-neighboring atoms of the ligands are in agreement with previous results on similar complexes [11]. The metal atoms in Co(II) and Ni(II) complexes have a distorted octahedral coordination sphere in which the two axial M-S bonds are elongated (Figure 3). The high-spin state of cobalt and nickel (2(S+1) = 4 for Co and 3 for Ni) is evidenced from the experimental magnetic susceptibility data (4.87 BM for Co and 3.12 BM for Ni). Complexation increased the bond length of C-S bond in both Ni and Co complex by 0.06 Å. The N-N bond length of oxadizaole (oxadiazole moiety complexing with metal atom) and the metal to M-X bond distances Ni and Co complexes are 2.63and 2.70 (S), 2.16 and 2.22 (N of pyridine moiety), 2.06 and 2.07 (O of water), respectively. An examination of frontier molecular orbitals reveals that in the case of Co(II) & Ni(II) complexes, the frontier occupied molecular orbitals have considerable metal orbital character (Figure 4); as expected from the crystal field theory which predicts that in a MLx complex, metal d- orbitals are pushed up to become frontier occupied orbitals, whereas the ligand orbitals are stabilized because of the orbital interactions between the metal and ligand.
IR Spectroscopic Characteristics The IR spectroscopic characteristics were used to identify the sites of coordination and evidence for the presence of lattice water. The free ligand shows prominent bands at 1556cm-1(υ(C-N) of oxadiazole ring). Computed value is 1557 cm-1. The υ(C-N) in the Ni and Co complexes shows a redshift and is observed at 1542 cm-1 and 1519 cm-1 (bonded to Ni), respectively. The redshift can be attributed to complexation with metal atom [25]. The computed values are matching with experimental observations and computed to be 1579 cm-1 and 1528 cm-1 for the Ni complex and 1542 cm-1 and 1533 cm-1 for the Co complex. A peak was observed at 1222 cm-1(1260 cm-1 theoretical) for the ligand, which can be assigned as δNH. In both the complexes, the frequency is redshifted by about 15 cm-1 and observed around 1207 cm-1. Computed value is about 1260 in both complexes, and may be due to the fact that computations are done on an isolated gas phase molecule. The ligand shows a vibrational frequency at 947 cm-1 (theoretical 994 cm-1), corresponding to the υ(N-N) vibration of the oxadiazole ring. In the metal complexes, these values also got redshifted and are about 933 cm-1 (computed values are around 967 cm-1 and 936 cm-1). These data suggest that the metal interacts with the ligand through the sulphur atom. The four bands of free ligand observed at around 1579 cm-1, 1556 cm-1, 1516 cm-1 and 1399 cm-1 have been assigned to C = C and C = N skeletal frequencies of the pyridine ring [11, 26]. Coordinating with metal ions, the peaks are shifted to 1587 cm-1 (1589 cm-1), 1567 cm-1 (1564 cm-1), 1519 cm-1 (1519 cm-1) and 1400 cm-1 (1402 cm-1) for the Co (Ni) complex. Computations show peaks at ~ 1579 cm-1 /1575 cm-1, 1557 cm-1 /1549 cm-1, 1511 cm-1 /1492 cm-1 and 1430 cm-1 /1437 cm-1 for pyrdine ring complexed and uncomplexed with the central metal atom. In the metal complex, a new band is observed around 431 in Co and 420 in Ni. Computations showed that these frequencies are corresponding to coordinated water molecule O-M vibrations, and computed values are around 413. A peak due to υPy was observed for the free ligand at 1603 cm-1 (theoretical 1623 cm-1) [26]. The corresponding frequencies observed for Ni and Co complexes are blue-shifted to 1620 cm-1 and 1612 cm-1, respectively. Computations indicate these frequencies are around ~1618 cm-1, in agreement with the experiment values. The C-H wagging frequency was observed at 1399 cm-1 for free ligand and the corresponding values for Co and Ni complexes observed around the region of 1392 cm-1,1352 cm-1 and 1402 cm-1, 1351 cm-1, respectively. The computational values are around ~1416 cm-1 and ~1356 cm-1, respectively. The frequency shifts indicate that the metal also complexes with ligand through the pyridine ring, as shown in the Figure 3. The broad band around 3500 cm-1 corresponds to OH stretching of coordinated water in both metal complexes [27]. The computed values are around 3691 cm-1 and 3704 cm-1 for the Ni and Co complexes. Although computations give frequencies to the corresponding complexed water molecule around 2210 cm-1 and 1639 cm-1 for the Ni complex and 1660 cm-1 and 1627 cm-1 for the Co complex, in experimental spectra such peaks are absent. The reason for the absence of these bands may be due to the presence of short strong hydrogen bonds, which enables shuttling of H between O of water and the ligand.
Electron Population Analysis To describe the anion-cation interactions and charge delocalization in detail, natural population analysis (NPA) at B3LYP level was carried out. Three types of metal-ligand interactions of particular interests are σ-donation by ligand lone pairs of electron, π-donation by ligands to the metal, and π-back-bonding from metal to ligand. Computed values of NPA are given in Table 3. A striking feature observed is that the NPA charge on the nitrogen and sulphur atoms directly bonded to metal cations has a higher negative charge than that in the free ligands. For example, in the cobalt complex, the negative charge on the nitrogen and sulphur atoms bonded to metal is increased by 0.22e and 0.45e units, respectively. This kind of localization of electron density in the ligand molecule on atoms directly bonded to metal ions can be understood in terms of the possibility of stronger electrostatic interaction and electron delocalization between the ligand and metal ion. The positive charge on the cation is less in Co(II) compared to Ni(II). The extent of electron transfer from ligand anion to metal cation could be evidenced from this observation. According to the study of Reed et al. [19], a change in population of 0.001e corresponds to the change of 0.627kcal mol-1 in energy of stabilization. The electron transfer occurring in the nickel complex computed to be 0.69e (NPA charge). From the charge delocalizing point of view, it is concluded that large ECT means strong charge delocalization from ligand atoms to cations, as well as less positive charges on cations. The NPA charges (Table 3) also supported this conclusion. The metal 3d-ligand orbital interaction, which contributes to covalent bonding in these complexes, becomes stronger in the Ni(II) than in the Co(II) complex, resulting from the increased effective nuclear charge of the metal atom in Ni(II) compared with Co(II) and the change in the 3d orbital populations. The distributions of electrons in these complexes, as determined by partial charges obtained using natural population analysis (NPA), are included in Table 3. The reduced charges on metal ions could be attributed to the nephelauxetic effect, in which the ligand charge donation partially shields the metal d electrons from the central ion nuclear charge and drives expansion of the d-electron "cloud". The identity of the metal [M (II) (M=Co, Ni)] was more clearly evidenced from the computed spin density for each complex. The excess spin on the metal was within 0.25 units of formal expectation for the respective system. The results are included in Table 4. The distribution of Mulliken spin density revealed the presence of significant spin delocalization onto the sulfur, oxygen and nitrogen atoms coordinating the metals. Most of the spin density was located within the first sphere of ligand atoms. This spin delocalization is due to the polarizability of sulfur ligands and the high spin configuration adopted by metals ions in the electronic ground state of the complexes.
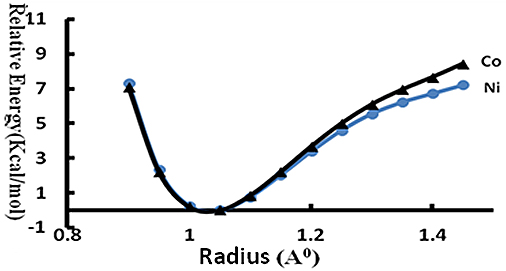
Evidence for Short-Strong Hydrogen Bonds in Co(II) / Ni(II) Complexes In natural photosynthetic systems, the short-strong hydrogen bond network with water functioned as a proton exit channel [17]. The spectroscopic characteristics and orbital characteristics of the Ni(II) and Co(II) complexes revealed that strong electrostatic interaction and electron delocalization between the ligands and the metal ions were present in these complexes, a critical requirement of photo-catalytic activity. In addition to this, if these complexes possess short-strong hydrogen bonds, then this system will be effective for photo-catalytic water splitting/artificial photosynthesis. The energy profile of H-transfer between water molecule and the ligand has been computed and presented in Figure 5. Transfer of the proton from the oxygen of the water molecule to the nitrogen atom of the ligand requires only <7 kcal/mol. This result revealed the presence of short-strong hydrogen bonds [28,29]. The distance between the two heteroatoms involved in this hydrogen bond is in the range of 2.5 Å, which is much shorter compared to an ordinary hydrogen bond where the corresponding distance is ~2.8 Å. The presence of short-strong hydrogen bonds and favorable electronic properties of the Co(II) / Ni(II) complexes of 5-(pyridin-4-yl)–3H-1, 3, 4–oxadiazole-2–thione revealed that the two complexes would mimic the natural leaf of plants that split water in to hydrogen and oxygen.
Conclusions The geometry of the synthesized Co(II) / Ni(II) complexes of 5-(pyridin-4-yl)–3H-1,3,4–oxadiazole-2-thione complexes were predicted as octahedral by considering the stoichiometry, magnetic moments and the results of electronic spectral analysis. Moreover the two tautomeric forms of the ligand 5-substituted-1,3,5-oxadiazole-2-thione was isolated along with the transition state using DFT modeling. The results of the DFT calculations revealed that the thione form is more stable at 10.3 kcal/mol compared to the thiol form. The transition state isolated using QST2 possesses an energy barrier of 34.6 kcal/mol. ESP computations revealed that there are two coordination sites in the 5-substituted-1,3,5-oxadiazole-2-thione ligand. Considering the results of ESP computations, electronic, IR spectroscopic characteristics and magnetic susceptibility analysis the Ni and Co complexes were modeled using DFT. Orbitals analysis revealed that in the case of Co(II) and Ni(II) complexes, the frontier occupied molecular orbitals have considerable metal orbital character. The NPA analysis revealed that strong electrostatic interaction and electron delocalization between the ligands and the metal ions were present in these complexes, a critical requirement of photo-catalytic activity. Moreover, the Ni(II) and Co(II) complex exhibited shortstrong hydrogen bonds similar to natural photosynthetic system. These results revealed that the synthesized complexes will be a potential candidate for the development of next generation artificial photosynthesis systems. References
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||